
Die Myelodysplastischen Syndrome (kurz MDS) umfassen eine Gruppe heterogener Erkrankungen der blutbildenden Stammzellen im Knochenmark (hämatopoetische Stammzellen), welche durch eine Verminderung von Blutzellen (Zytopenie) und das Vorhandensein von funktionsgestörten Zellen (Dysplasie) charakterisiert sind.
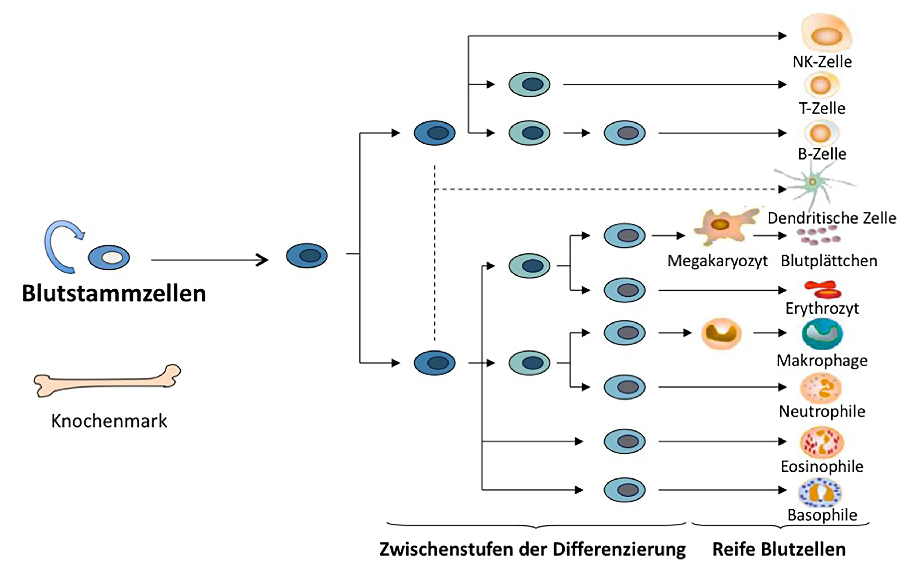
Hämatopoetische Stammzellen
Der menschliche Körper besitzt viele verschiedene Zelltypen, welche spezifischen Funktionen dienen. Nicht alle Zellen sind dabei in der Lage sich zu teilen, um beispielsweise bei einem Zelltod, die fehlenden Zellen wieder zu ersetzen. Ist eine Zelle in der Lage sich zu teilen, ist es notwendig, dass das Erbgut dabei ohne Fehler auf die Tochterzellen übertragen wird. Entsteht bei der Teilung jedoch ein Fehler, können Krebserkrankungen entstehen.
Stammzellen sind undifferenzierte Zellen, die sich selbst erneuern (Selbsterneuerung) und in unterschiedliche Zelltypen entwickeln können (Zelldifferenzierung). Das Potenzial kann dabei unterschiedlich sein und man spricht von Totipotenz (frühe embryonale Stammzellen), Pluripotenz (embryonale Stammzellen) und Multipotenz (fetale oder adulte Stammzellen):
Totipotente Stammzellen sind in der Lage durch Zellteilung ein komplett eigenständiges Individuum zu entwickeln. Diese Fähigkeit verlieren die Stammzellen bereits wenige Tage nach der Befruchtung (8-Zellstadium) und man spricht dann anschliessend von Pluripotenz.
Pluripotente Stammzellen sind in der Lage sich noch in alle embryonalen Gewebe (Entoderm, Mesoderm, Ektoderm) zu differenzieren.
Multipotente Stammzellen besitzen nur noch ein eingeschränktes Differenzierungspotential eines bestimmten Zelltyps. Sie sorgen in den verschiedenen Geweben bei Bedarf für Zellnachschub.
Die myeloische und lymphatische Zelllinien
Aus den hämatopoetischen Stammzellen entstehen myeloische und lymphatische Vorläuferzellen im Knochenmark, aus welchen sich die abschliessend differenzierten Zellen des Blutes entwickeln. Aus der myeloischen Vorläuferzelle entsteht ein Teil der weissen Blutkörperchen (Granulozyten und Monozyten), rote Blutkörperchen (Erythrozyten) und Blutplättchen (Thrombozyten). Aus den lymphatischen Vorläuferzellen hingegen entstehen Lymphozyten, welche den anderen Teil der weissen Blutkörperchen bilden. Die Lymphozyten lassen sich wiederum in drei Zelltypen, den B-, T- und NK-Zellen unterteilen.
Abbildung 1: Differenzierungsbaum der Blutzellbildung (Hämatopoese)¹
Zelluläre Bestandteile des Blutes
Die roten Blutkörperchen (Erythrozyten) dienen dem Sauerstofftransport und sind somit für die Energieversorgung des Körpers zuständig. Sie enthalten Hämoglobin, ein eisenhaltiger Eiweisskomplex, welcher Sauerstoff bindet und dem Blut die typisch rote Farbe verleiht. Besitzt der Körper zu wenige rote Blutkörperchen, und damit zu wenig Hämoglobin, bezeichnet man dies als Blutarmut (Anämie). Typische Anämiesymptome manifestieren sich mit (belastungsabhängiger) Atemnot (Belastungsdyspnoe), Müdigkeit, hohem Puls (Tachykardie), Blässe und Schwindel.
Die Blutplättchen (Thrombozyten) sind für die primäre Blutgerinnung zuständig. Sie sorgen dafür, dass blutende Wunden rasch gestillt werden und eine Wundheilung eingeleitet wird. Zu wenig Plättchen wird als Thrombozytopenie bezeichnet und manifestiert sich mit einer längeren Blutungsdauer sowie auch einem erhöhten Risiko für (Spontan-)Blutungen.
Die weissen Blutkörperchen (Leukozyten) dienen dem Körper zur Abwehr und bilden somit ein wichtiger Bestandteil unseres Immunsystems. Zu wenig weisse Blutzellen wird als Leukopenie bezeichnet und manifestiert sich mit einer erhöhten Infektanfälligkeit. Da die Leukozyten jedoch noch in weitere Zelltypen unterteilt werden (Lymphozyten, Monozyten und Granulozyten) ist je nach Herkunft der Zellen mit unterschiedlichen Konsequenzen für den Patienten zu rechnen. Hierbei ist bei einer Leukopenie eine Verminderung der Lymphozyten von derjenigen der Granulozyten oder auch Monozyten abzugrenzen.
Die Lymphozyten sind Teil der erworbenen Immunität und können in drei hauptsächliche Zelltypen eingeteilt werden
- B-Lymphozyten sind fähig Antikörper zu produzieren. Diese zirkulieren im Blut und können körperfremde Substanzen (Pathogene) erkennen und markieren. Diese Markierung unterstützt dann andere Immunzellen bei der Erkennung und Elimination der Pathogene. Da jedoch jedes Pathogen ein eigenes Erkennungsmerkmal besitzt, muss bei jeder neuen Infektion ein passender Antikörper gebildet werden.
Liegt ein passender Antikörper vor, können sich B-Lymphozyten auch in Gedächtniszellen differenzieren. Dies ermöglicht bei einem ähnlichen Pathogen, sich an dieses zu erinnern und von Beginn an passende Antikörper zu produzieren. Dadurch wird eine effizientere Immunantwort ausgelöst. - T-Lymphozyten können entweder Erreger direkt durch Zytokine abtöten (zytotoxische T-Zelle) oder durch Aktivierung von anderen Abwehrzellen die Immunantwort verstärken (T-Helferzelle). Diese Differenzierung findet jedoch erst nach Kontakt mit einem Pathogen statt, wodurch die T-Lymphozyten-Antwort mit einer leichten Verzögerung eintritt. Auch bei den T-Zellen bilden sich nach einer Immunantwort Gedächtniszellen
- NK-Zellen sind, wie zytotoxische T-Zellen, in der Lage Zellen direkt mittels Zytotoxine abzutöten. Im Gegensatz zu den T-Zellen benötigen NK-Zellen jedoch keine vorherige “Schulung” und sind somit die ersten aktiven lymphoiden Abwehrzellen. Eigentliche Gedächtniszellen gibt es aufgrund der fehlenden Schulung bei den NK-Zellen jedoch nicht.
Die Monozyten (Fresszellen) sind Teil der angeborenen Immunität und können ins Gewebe wandern, wo sie zu Gewebe-Makrophagen differenzieren. Dort fressen (phagozytieren) sie totes Zellmaterial, körperfremde Substanzen, Erreger (Viren, Bakterien, Parasiten und Pilze) sowie auch Immunkomplexe.
Die Granulozyten werden in 3 Zelltypen unterschieden.
- Neutrophile Granulozyten sind die “Frontsoldaten” der angeborenen Immunität und schützen uns vor Bakterien. Sie sind die am häufigsten vorhandenen Granulozyten und ein Mangel wird als Neutropenie bezeichnet. Eine Neutropenie ist mit einem erhöhten Risiko für bakterielle Infektionen assoziiert.
- Eosinophile Granulozyten dienen der Parasitenabwehr und spielen zudem bei Allergien eine Rolle.
- Basophile Granulozyten spielen v.a. bei Allergien eine Rolle. Sie sind die im Blut am seltensten vorkommenden Granulozyten.
Myelodysplastische Syndrome
Epidemiologie
In der Schweiz leben ca. 20/100’000 Personen (ca. 1680 Personen) mit der Diagnose eines MDS (Prävalenz).
Jedes Jahr erkranken 2-3/100’000 Personen (ca. 300-400 Personen) und davon ca. 25/100’000 Personen im Alter ≥65 Jahren an einem MDS. Dabei sterben jährlich ca. 150 Menschen an der Krankheit MDS.
Das mittlere Erkrankungsalter liegt über dem 70 Lebensjahr und stellt damit eine Erkrankungen des älteren Menschen dar.
Risikofaktoren
Folgende Faktoren erhöhen das Risiko an einem MDS zu erkranken:
- Arbeiten mit organischen Lösungen (Benzol), Insektiziden und Pestiziden
- Strahlenbelastung
- Chemotherapien
- Zunehmendes Alter
- Männliches Geschlecht
- Vererbte Prädispositionsyndrome (Fanconi-Anämie, Telomeropathien, Shwachman-Diamond Syndrom, etc.), v.a. zu beachten bei jüngeren MDS Patienten (Alter < 50 Jahren)
Symptome
Die MDS Erkrankungen sind durch Zytopenie und Dysplasie charakterisiert. Das Knochenmark ist oft paradoxerweise voll mit dysplastischen Zellen, die jedoch nicht gut ausreifen, früh absterben und somit nicht in die Peripherie ausgeschwemmt werden. Dies wird als ineffektive Blutbildung (ineffektive Hämatopoese) bezeichnet.
Bei den MDS können die drei verschiedenen Blutzellreihen unterschiedlich schwer von Zytopenie und Dysplasie betroffen sein. Für die Diagnose eines MDS muss in der Regel mindestens eine Zellreihe vermindert sein und im Mikroskop mindestens 10% der Zellen einer Reihe Dysplasien aufweisen.
Die Symptome können daher sehr vielfältig sein und da es sich oft um ältere Patienten handelt, kann die Zytopenie die Symptome anderer Erkrankungen zusätzlich verschlimmern.
Sind die roten Blutkörperchen vermindert, so leidet der Patient an Symptomen der Anämie. Der Körper wird nicht mehr genügend mit Sauerstoff versorgt und den Betroffenen fehlt es deswegen an Energie und sie fühlen sich nicht mehr leistungsfähig, müde und abgeschlagen.
Sind die weissen Blutkörperchen vermindert oder funktionsgestört, fehlt es dem Körper an funktionsfähigen Abwehrzellen. Deswegen sind die Betroffenen anfälliger für Infektionen. Bei einer schweren Neutropenie besteht ein erhöhtes Risiko für eine bakterielle Blutvergiftung, welche im schlimmsten Fall lebensbedrohlich werden kann.
Sind die Blutplättchen vermindert oder funktionsgestört, kann der Körper blutende Wunden nicht mehr stillen. Die Betroffenen bluten daher öfters und auch länger. Typisch hierfür sind kleinste punktförmige Einblutungen in der Haut der Beine und Füsse (Petechien), sowie Einblutungen in Haut und Schleimhaut (mukokutane Blutungen)
Diagnose
Die Diagnose der MDS wird mittels Blutbildes, Knochenmarks-Punktion, Zytogenetik und Gen-Sequenzierungs Methoden gestellt.
Knochenmark-Punktion
Da MDS durch Störungen der Blutbildung charakterisiert sind, braucht es hierfür die Untersuchung des blutbildenden Organs, dem Knochenmark. Dazu wird unter lokaler Betäubung und mit Hilfe einer Hohlnadel in den Knochen (meist im hinteren Beckenkamm) gestochen und ein Teil des blutbildenden Knochenmarkes entnommen. Hierzu wird dem Patienten oft auch ein Schmerz- und Schlafmittel abgegeben, da beim Ansaugen (Aspiration) des Markes ein schmerzhafter Unterdruck im Knochen erzeugt wird. Unter dem Mikroskop können anschliessend die entnommenen Zellen (Zytologie) nach Menge und Qualität (Dysplasie) in vielfältiger Weise untersucht werden. Die Biopsie wird entkalkt (Histologie) und ermöglicht eine bessere räumliche Beurteilung sowie den allfälligen Nachweis einer Faservermehrung (Fibrose) im Gewebe des Knochenmarkes. Beide Untersuchungen (Zytologie und Histologie) sind grundsätzlich notwendig, da sie unterschiedliche Aspekte des Knochenmarkes untersuchen.
Abbildung 2: Klassische Beispiele von Dysplasien der drei Zellreihen

links: Ringsideroblasten (dysplastische Vorläuferzellen der roten Blutkörperchen)
mitte: Pseudo-Pelger (dysplastische Neutrophile)
rechts: dysplastische Megakaryozyten (Vorläuferzellen der Blutplättchen)
Zytogenetik
Da MDS mit genetischen Veränderungen einhergehen, müssen Chromosomenveränderungen mittels Zytogenetik analysiert werden. Dabei werden die Chromosomen in der Zellteilungsphase gefärbt (Metaphasen Zytogenetik) und unter dem Mikroskop bezüglich struktureller und numerischer Veränderungen untersucht. Mit der konventionellen Metaphasen Zytogenetik lässt sich zum Beispiel der Verlust des langen Armes vom Chromosom 5 – die Deletion 5q – nachweisen, welches ein spezieller MDS Subtyp charakterisiert.
Gen Sequenzierung
Mit Hilfe von Sequenzierungsmethoden lässt sich die genaue DNA-Sequenz eines Genes bestimmen. Dadurch können DNA-Bausteinabweichungen (Basenveränderungen oder Mutationen) identifiziert werden. Diese Methode ist komplementär zu der Zytogenetik, da bei der groben Chromosomenanalyse die kleinen Verluste einer Genregion verpasst werden. Der Nachweis von Gen-Mutationen spielt eine zunehmend wichtige Rolle, nicht nur um das Verständnis der Erkrankung zu vertiefen, sondern auch um die Prognose besser abschätzen und gegebenfalls auch spezifische Ansatzpunkte (Targets) identifizieren zu können. Bereits jetzt wird die Next-Generation-Sequenzierung, im Rahmen von Panel-Sequenzierungen von 20-40 Genen, für Diagnostik, Prognose und Ansprechen auf Therapien zunehmend eingesetzt.
Klassifikation
Entsprechend der durchgeführten Diagnostik und der Anzahl betroffenen Zellreihen nach Dysplasie und Zytopenie werden die MDS, nach WHO-Klassifikation 2016, in Subtypen eingeteilt.
Prognose
Die Prognose des MDS variiert je nach Ausmass der Zytopenien, Vorliegen von unreifen Zellen (Blasten) und genetischen Veränderungen stark (krankheitsspezifische Faktoren). Auch die Vorerkrankungen des Patienten spielen für die Prognose eine entscheidende Rolle (patientenspezifische Faktoren). Das Spektrum der natürlichen Verläufe umfasst dabei einen langsamen, stabilen Zustand über mehrere Jahre bis zu einem raschen Übergang in eine lebensbedrohliche Akute Myeloische Leukämie (AML), welche durch einen Anstieg der Blasten ≥ 20% im peripheren Blut oder Knochenmark definiert ist.
Um die Prognose abschätzen und eine angepasste Therapie einleiten zu können, wurden daher verschiedene krankheitsspezifische Prognosesysteme wie z.B. den Revised International Prognostic Scoring System (IPSS-R) entwickelt. Dieser berücksichtigt für die Prognoseneinschätzung Anzahl/Schweregrad der Zytopenien, Anzahl Blasten und zytogenetische Veränderungen.
Nebst dem krankheitsspezifischen Prognosesystem finden auch patientenspezifische Prognosesysteme ihre Anwendung. Der Hematopoietic Cell Transplantation-specific Comorbidity Index (HCT-CI) schätzt anhand möglicher Vorerkrankungen eines Patienten das Risiko und die daraus folgende Sterblichkeit bei einer geplanten Stammzelltransplantation ab.
Als vereinfachte Form des patientenspezifischen Screenings wird der MDS-Comorbidity Index (MDS-CI) verwendet. Dieser basiert auf einer kleineren Anzahl an möglichen Vorerkrankungen, sodass dieser schnell und einfach anzuwenden ist. Der MDS-CI wird daher bei eher älteren MDS-Patienten eingesetzt, die sich nicht für eine Stammzelltransplantation qualifizieren.
Therapie
Die Behandlung richtet sich primär nach krankheits- (IPSS-R) und patientenspezifischen Faktoren.
- Abnahme der bereits verminderten Zellanzahl mit entsprechender klinischer Symptomatik
- Zunehmender Transfusionsbedarf
- Zunehmende Anzahl unreifer Zellen im peripheren Blut und/oder Knochenmark (Blasten)
- Nachweis zusätzlicher zytogenetischer (evtl. molekularer) Abweichungen
- Fehlendes Ansprechen auf eine Therapie
- schwere, unkontrollierte immunologische Begleitphänomene (Autoinflammation, Autoimmunität)
Niedrigrisiko MDS (IPSS-R: sehr niedrig , niedrig und intermediär)
Bei Betroffenen mit einem niedrigen Risikoscore wird nur bei bestehenden Symptomen einer Anämie (Thrombopenie und Neutropenie häufiger bei Hochrisiko Patienten) behandelt. Diese hat das primäre Ziel, die Lebensqualität des Patienten zu verbessern (supportive Therapie). Bei speziellen MDS Formen kommen auch krankheitsmodifizierende Behandlungen in Frage, welche den natürlichen Verlauf der Erkrankung günstig beeinflussen können.
Folgende Therapien kommen zum Einsatz:
Transfusionen
Blut-Konzentrate (Konzentrate von roten Blutkörperchen oder Erythrozyten) können bei symptomatischer Blutarmut (Anämie) zum Einsatz kommen. Dadurch können die roten Blutzellen erhöht und somie die Symptome der Anämie gelindert werden.
Erythrozyten besitzen spezifische Oberflächenstrukturen, welche für jeden Menschen einzigartig sind und auch die Blutgruppe definieren. Bei der Gabe von Erythrozyten-Konzentraten besteht deswegen das Risiko, sich fremde Oberflächenstrukturen (Antigene) zu immunisieren, welche bei einer erneuten Transfusion schwerwiegende Komplikationen verursachen können (Allo-Antikörper mit Hämolyse). Dieses Risiko wird durch die vorgängige Analyse der Blutgruppeneigenschaften und vorliegenden Antikörperkonstellation (Type and Screen) sowie der Auswahl von geeigneten Blutkonserven minimiert. Weiter enthalten Blutprodukte sehr viel Eisen, welches bei häufigen Transfusionen zu einer Eisenüberladung des Körpers führen kann (siehe Eisenchelationstherapie). Und zuletzt kann eine Transfusion bei Patienten mit Herz- und Nierenerkrankungen zu einer Volumenüberladung führen.
Auch Thrombozyten-Konzentrate (Konzentrate von Blutplättchen) können bei einem Thrombozytenmangel zugeführt werden.
Dadurch werden die Werte der Blutplättchen verbessert, wodurch eine Blutung gestillt und das Blutungsrisiko gesenkt wird. Ähnlich wie bei den roten Blutzellen, können sich Patienten auch gegen fremde Oberflächeneigenschaften der Plättchen immunisieren (HLA/HPA-Antikörper), zudem treten selten auch unspezifische immunologische Reaktionen auf. Mit einem Bestrahlungsverfahren werden heute in allen Thrombozyten-Konzentraten mögliche Pathogene (Bakterien, Viren) unschädlich gemacht.
Da mit den Blutbestandteilen auch Leukozyten und Serum transfundiert werden, kann es zu infektiologischen (Hepatitis und HIV) und immunologischen Komplikationen (Fieber, Entzündungsreaktionen) kommen. Durch eine rigorose Spenderwahl und Bestrahlung der Blutprodukte wurden diese Risiken in den letzten Jahren jedoch erheblich vermindert, können aber nie vollständig ausgeschlossen werden.
Eisenchelationstherapie
Erythrozyten beinhalten viel Eisen, welches der Körper alleine nicht genügend ausscheiden kann. Da MDS-Patienten oft mehrere Blut-Transfusionen erhalten, droht dem Patienten daher eine Eisenüberladung, welche auf Dauer den Körper erheblich belastet. Das überschüssige Eisen lagert sich in Leber, Herz und anderen Organen ab und kann zu einer Leberzirrhose, Herzinsuffizienz und anderen Beschwerden führen. Mit einer Eisenchelationstherapie lässt sich das freie Eisen im Blut abbinden, was zu einer vermehrten Eisenausscheidung führt.
Wachstumsfaktoren
Nebst der Blut-Transfusion und Eisenchelation kann auch die Blutbildung durch eine medikamentöse Behandlung verbessert werden.
Erythropoetin (EPO) aktiviert die Bildung von roten Blutkörperchen, Thrombopoetin diejenige von Blutplättchen, und Granulozyten-Wachstumsfaktoren die Bildung von neutrophilen Granulozyten.
Infektprophylaxe
Um die erhöhte Infektionsgefahr gegenüber Bakterien, Viren oder Pilzen abzuwenden, kommen zum Teil auch anti-infektiöse Prophylaxen zum Einsatz. Diese müssen jedoch sehr gezielt und bewusst eingesetzt werden, um nicht die Entwicklung von Resistenzen zu fördern.
Krankheitsmodifizierende Therapie (bei speziellen MDS Formen)
Bei gewissen MDS-Formen existieren zusätzliche Therapiemassnahmen, welche die Bildung von reifen Blutzellen unterstützen und dadurch die Transfusionsabhängigkeit minimieren können.
Immuntherapie (ATG, CyA)
Antithymozytenglobulin (ATG) und Cyclosporin A (CyA) sind beides immunsupprimierende Medikamente (Immunsuppressiva). Dies bedeutet, dass sie Einfluss auf das Immunsystem nehmen und dieses hemmen. Bei einem hypoplastischen MDS (Knochenmark mit wenigen Zellen) mit niedrigem Risiko (IPSS-R ≤ 3) konnte gezeigt werden, dass dadurch die Zahl der funktionsfähigen Blutzellen wieder ansteigt. In 30% der Patienten konnte sogar eine Transfusionsfreiheit erreicht werden.
Da ATG aus anderen Tieren (Pferde, Hasen) gewonnen wird und dabei auch starken Einfluss auf unser Immunsystem nimmt, kann dieses teilweise schwere Nebenwirkungen (z.B. einen allergischen Schock) auslösen, sodass dieses Medikament nur in einem hämatologischen Zentrum angewendet werden soll.
Lenalidomid
Lenalidomid gehört zu den immunmodulierenden Medikamenten. Dies bedeutet, dass es Einfluss auf das Immunsystem nimmt, dieses jedoch stellenweise hemmt sowie auch stellenweise aktiviert. Patienten mit einer einzelnen Chromosom 5q Deletion (MDS del 5q) bei einem Niedrig-Risiko MDS (IPSS-R ≤ 3) erreichten in 60% der Fälle Transfusionsfreiheit für 1-2 Jahre. Ähnlich gut sprechen zudem Patienten mit einer zusätzlichen Chromosomenabweichung (ausser vom Chromosom 7) an.
Luspatercept
Luspatercept ist ein Medikament, welches die Differenzierung und die Proliferation der roten Blutzellen fördert (Erythroid Maturating Agent). Es zeigt vor allem bei Patienten mit einem MDS mit Ringsideroblasten Wirkung, sodass ca. 60% dieser Patienten eine Verbesserung der Anämie mit Reduktion der Transfusionsbedürftigkeit und in 30% eine Transfusionsfreiheit erreichen können. Luspatercept ist zur Zeit nur bei transfusionsabhängigen Patienten zugelassen, die vorher nicht auf eine Therapie mit Erythropoietin angesprochen haben oder die Wahrscheinlichkeit des Ansprechens gering ist.
Hochrisiko MDS (IPSS-R: hoch und sehr hoch)
Bei Betroffenen mit einem Hochrisiko MDS sind Therapien notwendig, welche nicht nur die Befindlichkeit verbessern (supportive Therapie) sondern auch den Krankheitsverlauf günstig beeinflussen können (krankheitsmodifizierende Behandlung). Nebst der supportiven Therapie (siehe niedriges Risiko) kommen folgende Therapien zum Einsatz:
Hypomethylierende Substanzen (z.B. Azacytidin, Decitabine)
Azacytidin und Decitabine sind Substanzen, welche einer der DNA-Basen sehr ähnlich sind (Cytidin-Analogon) und bei der Zellteilung in die DNA eingebaut werden. Dies hat zur Folge, dass die genetische Information aus der DNA anders abgelesen wird und auf diese Weise die Zellteilung und Differenzierung günstig beeinflusst werden. Dadurch reifen die unreifen Blutzellen besser aus, gefolgt von einer Zunahme an funktionsfähigen Zellen im Blut. Die genetischen Veränderungen selber, werden aber durch diese Therapie nicht beeinflusst und bei allen Patienten nimmt die Wirkung mit der Zeit ab.
Chemotherapie
Die klassischen Chemotherapeutika sind Arzneimittel, welche die Zellteilung erheblich hemmen und dadurch die bösartigen Zellen abtöten. Die Chemotherapeutika wirken jedoch unspezifisch und somit auch auf normale Zellen, die auch eliminiert werden. Daher können Chemotherapeutika zu einem vorübergehenden Abfall aller Blutzellen (Aplasie) und anderen Nebenwirkungen (Haarausfall, Durchfall, Schleimhautschäden) führen. Das Ziel der Chemotherapie ist es, die bösartigen Zellen zu verdrängen und den gutartigen Zellen in der Erholungsphase der Knochenmarksfunktion einen Vorteil zu verschaffen. Da beim MDS die Stammzelle erkrankt ist, erholen sich die MDS Patienten oft nur zögerlich nach einer Chemotherapie und ohne nachfolgende hämatopoetische Stammzelltransplantation ist eine komplette Erholung der Blutwerte nur selten möglich und ein Rückfall der MDS Erkrankung meist unausweichlich.
Hämatopoetische Stammzelltransplantation (nur für “fitte” Patienten)
Die einzige Behandlung, welche die Patienten mit Hochrisiko-MDS heilen kann, ist ein Ersatz der kranken hämatopoetischen Stammzellen. Dies erfolgt durch eine vorbereitende Chemotherapie (Konditionierung), welche Raum für die anschliessende Transplantation von immunologisch kompatiblen, hämatopoetischen Stammzellen von einem gesunden Familien- oder Fremdspender (allo HSCT: allogene hämatopoetische Stammzelltransplantation) schafft. Nur wenige Patienten qualifizieren sich aufgrund des fortgeschrittenen Alters und der Begleiterkrankungen für eine solche, intensive Behandlung (ca 10% aller MDS Patienten). Das Risiko einer allogenen HSCT liegt hauptsächlich in der Toxizität der Konditierungschemotherapie sowie der umgekehrten Abstossung (GvHD: Graft vs. Host Disease). Bei der GvHD erkennen die transplantierten Immunzellen des Spenders Zellen im Körper des Empfängers als fremd, wodurch es zu lebensbedrohlichen, immunlogischen Reaktionen kommen kann. Diese Reaktion kann durch eine immununterdrückende Behandlung (Immunsuppression) vermieden und kontrolliert werden. Eine allo HSCT hat den Vorteil, dass dem erkrankten Empfänger funktionsfähige Blutstammzellen und Immunzellen eines gesunden Spenders zugeführt werden. Dadurch ist der Betroffene selbst wieder in der Lage, genügend funktionsfähige Blut- und Immunzellen zu produzieren, was einerseits die kranken Blutstammzellen durch gesunde ersetzt und andererseits das Immunsystem so verändert, dass die rechlichen “Krebszellen” erkannt und bekämpft werden. Nur diese Kombination von ersetzenden, gesunden Stammzellen und Immuntherapie gegen die kranken Stammzellen kann schlussendlich zur Heilung des MDS führen.
Erfahre hier mehr zur Blutstammzelltransplantation